سندرم واردنبرگ یک اختلال ژنتیکی نادر اما قابل شناسایی است که عمدتاً با کمشنوایی مادرزادی و تغییرات پیگمانتاسیون در پوست، مو و چشمها شناخته میشود. این سندرم برای نخستینبار توسط پزشک هلندی، Petrus Johannes Waardenburg، در سال ۱۹۵۱ توصیف شد. با وجود نادر بودن، تشخیص بهموقع آن برای پیشگیری از مشکلات شنوایی و کمک به رشد اجتماعی و زبانی بیمار اهمیت دارد. در این مقاله از سایت دکتر ابوئی به بررسی این موضوع میپردازیم. در ادامه همراه ما باشید.
اپیدمیولوژی
شیوع سندرم واردنبرگ در جوامع مختلف متفاوت است، اما تخمین زده میشود که از هر ۴۰,۰۰۰ نفر یک نفر به نوعی از این سندرم مبتلا باشد. حدود ۲ تا ۵ درصد از موارد ناشنوایی مادرزادی غیرسندرومی، ناشی از سندرم واردنبرگ هستند.
علائم و نشانهها
علائم بالینی سندرم واردنبرگ متغیر است و شدت آن میتواند از فردی به فرد دیگر حتی در یک خانواده متفاوت باشد. مهمترین ویژگیها عبارتند از:
شنواییپریشی (Hearing Loss)
معمولاً دوطرفه، مادرزادی و حسی-عصبی است.
در برخی موارد کامل (ناشنوایی مطلق) و در برخی دیگر جزئی است.
تغییرات پیگمانتاسیون
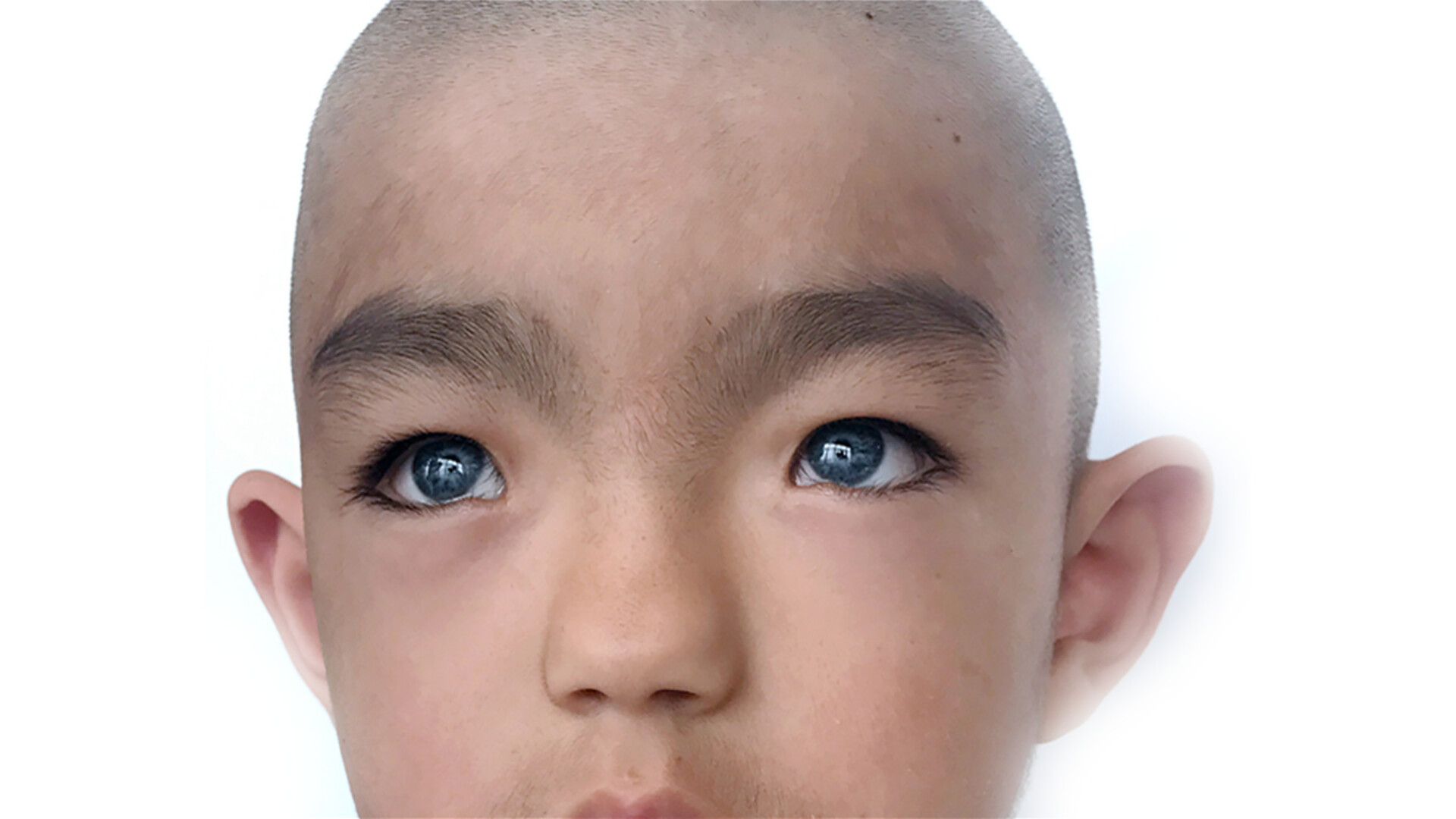
سفیدی موی ناحیه پیشانی (White Forelock)
چشمهای بسیار آبی یا هتروکرومی (دو رنگی چشمها)
لکههای بیرنگ روی پوست
موی سفید یا خاکستری در سنین پایین
ناهنجاریهای چهرهای
در نوع ۱، افزایش فاصله بین گوشههای داخلی چشمها (Dystopia canthorum)
ابروهای پهن و به هم پیوسته (Synophrys)
اختلالات سیستم گوارشی (در نوع ۴)
بیماری هیرشپرونگ: اختلال در حرکات روده ناشی از عدم وجود سلولهای عصبی در بخشی از کولون
اختلالات عضلانی-اسکلتی (نوع ۳)
ناهنجاریهایی در اندام فوقانی مانند انگشتان یا مفاصل غیرطبیعی
طبقهبندی انواع سندرم واردنبرگ
برای مشاهده بهتر جدول در گوشی همراه آن را به حالت افقی بگیرید.
نوع
ویژگیهای اصلی
ژن درگیر
الگوی وراثت
نوع ۱
فاصله زیاد چشمها + ناشنوایی
PAX3
اتوزوم غالب
نوع ۲
بدون فاصله چشم، دارای ناشنوایی
MITF، SOX10، SNAI2
اتوزوم غالب
نوع ۳ (Klein)
نوع ۱ + ناهنجاری اندامها
PAX3
اتوزوم غالب
نوع ۴ (Shah)
نوع ۲ + بیماری هیرشپرونگ
EDNRB، EDN3، SOX10
اتوزوم مغلوب
علل ژنتیکی
سندرم واردنبرگ حاصل جهش در ژنهایی است که در تشکیل و عملکرد سلولهای ملانوسیت نقش دارند. ملانوسیتها نه تنها مسئول تولید رنگدانه هستند، بلکه در گوش داخلی نیز نقشی حیاتی در انتقال صدا دارند. ژنهای مهم شامل:
PAX3 (شایع در نوع ۱ و ۳)
MITF (در نوع ۲)
SOX10، EDNRB، EDN3 (در نوع ۲ و ۴)
نحوه تشخیص
تشخیص بر اساس ترکیبی از علائم بالینی و آزمایشهای ژنتیکی صورت میگیرد:
معاینه فیزیکی کامل
ارزیابی شنوایی (Audiometry)
بررسی رنگ چشم، مو و پوست
آزمایش ژنتیکی جهت شناسایی جهش ژنی
در نوع ۴، تصویربرداری و بیوپسی روده برای بررسی هیرشپرونگ
درمان قطعی برای سندرم واردنبرگ وجود ندارد، اما مدیریت علائم بسیار مؤثر است:
استفاده از سمعک یا کاشت حلزون برای کمشنوایی
مشاوره ژنتیک برای خانوادهها
درمانهای جراحی برای بیماری هیرشپرونگ (در نوع ۴)
پشتیبانی روانی و آموزشی برای رشد گفتار و مهارتهای اجتماعی
پیشآگهی
بیشتر افراد مبتلا، بهویژه در صورت مدیریت مناسب شنوایی و گفتار، زندگی طبیعی و طول عمر عادی دارند. کیفیت زندگی بیماران به شدت علائم و میزان پشتیبانی بستگی دارد.
نتیجهگیری
سندرم واردنبرگ گرچه نادر است، اما با تشخیص بهموقع و مدیریت بینرشتهای (ژنتیک، گوشحلقبینی، روانشناسی و گفتاردرمانی) میتوان کیفیت زندگی بیماران را به طرز چشمگیری بهبود داد. افزایش آگاهی عمومی و تخصصی نسبت به علائم این سندرم، گامی مهم در مسیر تشخیص زودهنگام و پیشگیری از عوارض ثانویه است.
سندرم کورنیلا د لانگ (Cornelia de Lange Syndrome – CdLS) یک اختلال ژنتیکی نادر است که رشد فیزیکی، ذهنی و رفتاری را تحت تأثیر قرار میدهد. این سندرم نخستین بار توسط دکتر کورنیلا د لانگ، متخصص کودکان هلندی، در سال ۱۹۳۳ توصیف شد. میزان بروز آن تخمین زده میشود که بین ۱ در هر ۱۰٬۰۰۰ تا ۳۰٬۰۰۰ تولد زنده باشد. در این مقاله از سایت دکتر ابوئی به سندرم سندرم کورنیلا د لانگ میپردازیم، در ادامه همراه ما باشید.
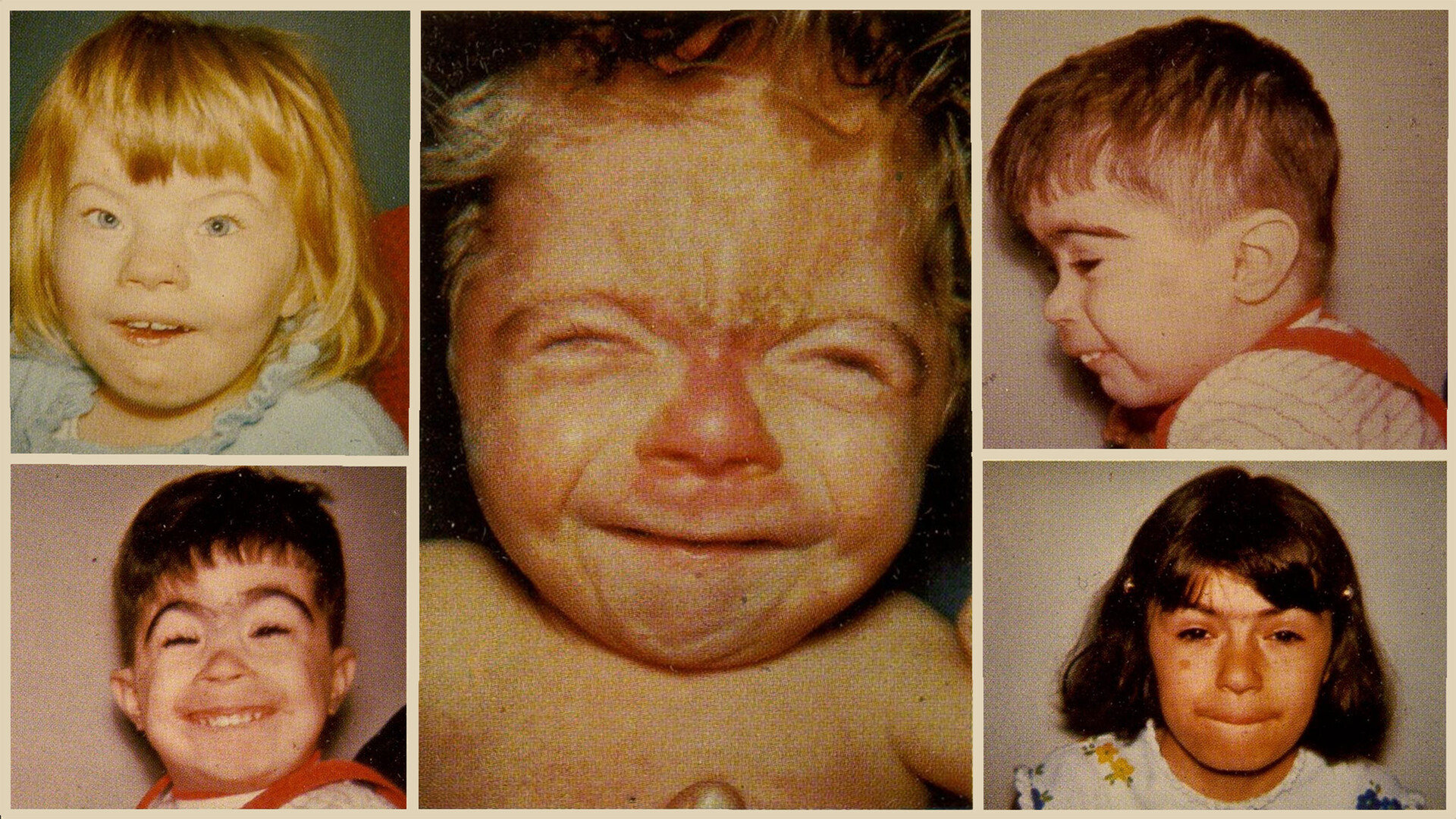
سینوفریز (Synophrys): بههم پیوستن ابروها در خط میانی.
مژههای بلند و ضخیم.
بینی کوتاه با سوراخهای بینی به سمت پایین.
لب بالایی نازک، لب پایینی برجسته.
فک پایین کوچک (Micrognathia).
گوشهای پایینتر از محل معمول و فرم غیرطبیعی.
تأخیر رشد:
رشد داخل رحمی (Intrauterine Growth Retardation).
قد و وزن کمتر از صدکهای طبیعی بعد از تولد.
ناهنجاریهای اندام:
کوتاهی اندام فوقانی (مخصوصاً بازو یا ساعد).
انگشتان اضافی (Polydactyly) یا فقدان انگشت (Oligodactyly).
مفاصل سفت یا محدودیت حرکتی.
اختلالات ذهنی و رفتاری:
تأخیر در گفتار.
درجات مختلف عقبماندگی ذهنی (از خفیف تا شدید).
رفتارهای اوتیسممانند (Autistic-like behaviors).
مشکلات سیستمیک:
ناهنجاریهای قلبی (مانند نقص دیواره بین بطنی – VSD).
ناهنجاریهای کلیوی.
رفلاکس معدهبهمری (GERD).
مشکلات شنوایی (کمشنوایی انتقالی یا عصبی-حسی).
مشکلات بینایی (مانند نزدیکبینی یا تنبلی چشم).
علتشناسی (Etiology)
CdLS ناشی از جهش در ژنهایی است که در چسبندگی کروماتین و تنظیم رونویسی ژنتیکی دخیل هستند:
جهت مشاهده بهتر جدول در گوشی همراه، آن را به حالت افقی نگه دارید
ژن درگیر
ویژگی
نوع وراثت
NIPBL (بیشتر موارد)
پروتئینی که در کمپلکس Cohesin نقش دارد
اتوزومال غالب
SMC1A
کروموزوم X
وابسته به X
SMC3
کمپلکس Cohesin
اتوزومال غالب
RAD21
ارتباط با Cohesin
اتوزومال غالب
HDAC8
کنترل ساختار کروماتین
وابسته به X
در اکثر موارد، این جهشها de novo هستند (یعنی به صورت جدید در فرد ایجاد شدهاند و از والدین به ارث نرسیدهاند).
تشخیص سندرم کورنیلا د لانگ (Diagnosis)
تشخیص مبتنی بر سه محور است:
معاینه بالینی
بر اساس ظاهر فیزیکی (صورت مشخصه، ناهنجاریهای اندام، تأخیر رشد).
آزمایشهای ژنتیکی
توالییابی نسل جدید (NGS) یا آزمایش پنل ژنتیکی برای شناسایی جهشهای NIPBL، SMC1A، SMC3، RAD21 و HDAC8.
در صورت شک بالینی قوی اما تست ژنتیک منفی، میتوان از آزمایشهای MLPA یا Array CGH برای کشف حذفهای کوچک (microdeletions) استفاده کرد.
سایر ارزیابیهای تکمیلی
اکوکاردیوگرافی برای بررسی مشکلات قلبی.
بررسیهای شنوایی و بینایی.
تصویربرداری MRI مغز در صورت وجود اختلالات عصبی.
پیشگیری از سندرم کورنیلا د لانگ (Prevention)
مشاوره ژنتیک (Genetic Counseling)
اگرچه بیشتر موارد CdLS تصادفی هستند، در مواردی که والدین حامل جهش باشند، ریسک تکرار وجود دارد.
آزمایش ژنتیکی والدین در صورتی که فرزندی با CdLS داشته باشند توصیه میشود.
غربالگری پیش از تولد
در خانوادههای با سابقه CdLS و جهش شناخته شده، میتوان با آمنیوسنتز یا نمونهبرداری از پرزهای کوریونی (CVS)، DNA جنین را آزمایش کرد.
در سونوگرافیهای دقیق (Anomaly Scan)، نشانههایی مانند تأخیر رشد جنینی، ناهنجاری اندام فوقانی و ویژگیهای غیرطبیعی صورت ممکن است قابل تشخیص باشند، ولی قطعی نیستند.
درمان قطعی برای CdLS وجود ندارد. درمان بر مدیریت علائم متمرکز است:
مراقبتهای پزشکی:
درمان مشکلات گوارشی (مانند GERD با دارو یا جراحی).
مداخلات شنوایی (سمعک یا جراحی).
اصلاح ناهنجاریهای ارتوپدی در صورت نیاز.
توانبخشی:
گفتاردرمانی، کاردرمانی و فیزیوتراپی برای ارتقای مهارتهای حرکتی و ارتباطی.
درمانهای روانشناختی و رفتاری به خصوص برای مشکلات اوتیسمی و پرخاشگری.
مداخلات آموزشی:
آموزشهای ویژه متناسب با سطح یادگیری کودک.
پشتیبانی در محیطهای مدرسه با استفاده از مربیان آموزشدیده.
پیشآگهی (Prognosis)
بسته به شدت علائم، پیشآگهی متفاوت است. در موارد خفیفتر، افراد میتوانند زندگی نسبتاً مستقلی داشته باشند، در حالی که در موارد شدید، نیاز به مراقبتهای مادامالعمر دارند. امید به زندگی معمولاً کاهش نمییابد مگر در صورت وجود مشکلات شدید قلبی یا ریوی درماننشده.
جمعبندی
سندرم کورنیلا د لانگ یک اختلال چندسیستمی با طیف بالینی گسترده است که نیازمند تشخیص زودهنگام، مراقبت چندتخصصی، و پشتیبانی خانوادگی قوی است. با پیشرفتهای ژنتیکی در شناسایی و تشخیص، امکان غربالگری قبل از تولد نیز فراهم شده، هرچند پیشگیری از جهش de novo هنوز ممکن نیست.
سندرم اهلرز-دانلوس (EDS) به عنوان گروهی ناهمگون از اختلالات ژنتیکی نادر شناخته میشود که بافت همبند بدن را تحت تأثیر قرار میدهد. این بیماری عمدتاً ناشی از نقص در ساختار، تولید یا پردازش کلاژن است. کلاژن، پروتئین اصلی مسئول ایجاد استحکام و انعطافپذیری در پوست، مفاصل، رگها و اندامها به شمار میرود. این سندرم به ۱۳ زیرگونه تقسیم میشود که هر کدام ویژگیها و عوارض خاص خود را دارند. در این مقاله از سایت دکتر ابوئی، اطلاعات جامع مرتبط با علل، روشهای تشخیص، درمان و مدیریت این سندرم (Ehlers-Danlos Syndromes) ارائه شده است.
انواع اصلی سندرم اهلرز-دانلوس
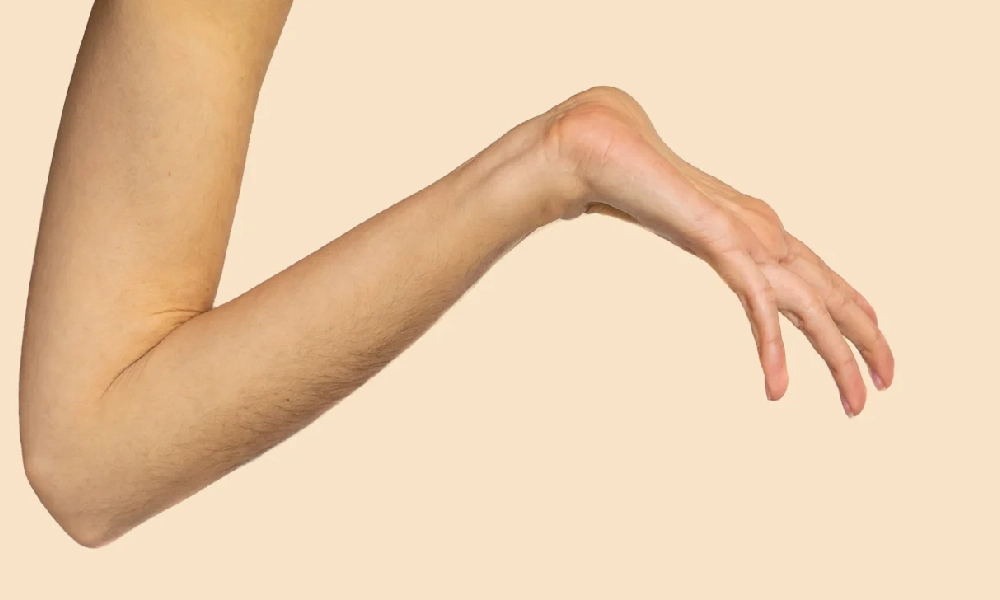
نوع هایپرموبایل (Hypermobile EDS یا hEDS):
شایعترین نوع، با شیوع حدود ۹۰ درصد موارد.
ویژگیها: مفاصل بیشازحد انعطافپذیر، درد مزمن مفاصل و پوست نرم و مخملی.
خطرات: دررفتگی مکرر مفاصل و احتمال بروز اختلالات خودایمنی همراه (مانند سندرم خستگی مزمن یا فیبرومیالژیا).
نوع کلاسیک (Classic EDS یا cEDS):
پوست بسیار کشسان با زخمهای گسترده همراه با اسکارهای نازک (مانند زخمهای “کاغذی”).
خطر پارگی پوست حتی در اثر ضربات جزئی و سایش سطحی.
نوع عروقی (Vascular EDS یا vEDS):
کشندهترین نوع، به دلیل ضعف قابلتوجه در دیواره رگهای خونی و اندامهای داخلی.
علائم بحرانی: پارگی ناگهانی عروق بزرگ (مانند آئورت) یا سوراخ شدن ناگهانی اندامها نظیر روده یا رحم.
انواع نادر:
شامل کیفواسکولار، آرتروکالازیا، درماتوسپاراکسیس و دیگر انواع خاص با شیوع کمتر.
معیارهای تشخیصی EDS برای هر زیرگونه شامل مجموعهای از علائم اصلی و فرعی است.
مثال: تشخیص نوع هایپرموبایل (hEDS) نیازمند وجود علائم بالینی مانند انعطافپذیری بالای مفاصل همراه با رد سایر اختلالات مشابه است. مقیاس بیگتون (Beighton Score) نیز بهعنوان یکی از ابزارهای ارزیابی مورد استفاده قرار میگیرد.
آزمایشهای ژنتیکی:
برخی انواع خاص EDS، نظیر نوع عروقی (vEDS)، با جهشهای ژنی مشخص مرتبط هستند (مانند جهش ژن COL3A1). آزمایش ژنتیک میتواند تأیید تشخیص را امکانپذیر کند.
روشهای تصویربرداری و آزمایشهای مکمل:
اکوکاردیوگرافی: برای شناسایی آنوریسم آئورت.
بیوپسی پوست: ارزیابی کلاژن در بیماران مشکوک به انواع نادر.
آزمایشهای خون: بررسی برخی بیماریهای همراه مثل لوپوس.
سندرم اهلرز-دانلوس (Ehlers-Danlos Syndromes) یکی از پیچیدهترین اختلالات ژنتیکی محسوب میشود که نیازمند درمان چندرشتهای و مدیریت دقیق است. با اتخاذ روشهای پیشگیری و کنترل، بیماران میتوانند به زندگی فعال و کیفیت مطلوب دست یابند. از زمانی که برای مطالعه این مقاله اختصاص دادید ممنونیم. لطفا با ارائه نظرات خود تیم تحریریه سایت دکتر ابوئی را در ارائه خدمات بهتر یاری کنید.
آنا ویسلز ویلیامز؛ بانویی در دل آزمایشگاه، پیشگامی در میدان نبرد با بیماریها. در سحرگاه قرن بیستم، زمانی که علم پزشکی هنوز درگیر مبارزهای نابرابر با بیماریهای کشندهای مانند دیفتری و هاری بود، زنی بیادعا و استوار وارد صحنه شد؛ زنی که صدای پایش در راهروهای تاریک آزمایشگاهها پیچید و رد پایش بر دیوارهای علم باقی ماند. این زن کسی نبود جز آنا ویسلز ویلیامز؛ باکتریشناس و پزشک آمریکایی که علم را نه تنها آموخت، بلکه با آن آیندهای روشنتر برای بشر رقم زد. در این مقاله از سایت دکتر ابوئی به بررسی زندگی این بانوی دانشمند میپردازیم.
از اندوه خانوادگی تا عزم راسخ برای خدمت
آنا در سال ۱۸۶۳ در شهر کوچک هکنساک در ایالت نیوجرسی به دنیا آمد. دوران کودکیاش همچون بسیاری از زنان آن عصر، در سایه محدودیتها گذشت، اما مرگ نوزاد خواهرش که از بیماری درمانناپذیر دیفتری جان باخت، آتش عجیبی در درون آنا روشن کرد. او در دل سوگ، تصمیم گرفت که زندگیاش را وقف شناخت بیماریها و مبارزه با آنها کند.
زن، میکروسکوپ و امید
در روزگاری که زنان کمتر به دنیای علم راه مییافتند، آنا ویسلز ویلیامز با پشتکاری بینظیر وارد کالج پزشکی زنان نیویورک شد و در سال ۱۸۹۱ فارغالتحصیل گردید. سپس راهی اروپا شد و در دانشگاههای معتبر وین، هایدلبرگ و لایپزیگ به تحصیل و پژوهش پرداخت.
اما نقطه عطف زندگی علمیاش، بازگشت به نیویورک و پیوستن به آزمایشگاه معروف بهداشت عمومی در کنار دکتر ویلیام پارک بود. آنجا بود که آنا تبدیل به ستونی در علم باکتریشناسی شد.
جنگ با دیفتری: تولد آنتیتوکسین نجاتبخش
در سال ۱۸۹۴، آنا ویسلز ویلیامز موفق شد نوعی باکتری دیفتری را جداسازی کند که بعداً با نام “پارک-ویلیامز شماره ۸” معروف شد. این کشف بیصدا اما عظیم، راه را برای تولید گسترده و ارزان آنتیتوکسین دیفتری باز کرد؛ درمانی که جان هزاران کودک را نجات داد.
در عصری که مرگ کودکان از دیفتری امری رایج بود، این موفقیت نه فقط علمی، بلکه انسانی و انقلابی بود.
آنا همچنین در توسعه روشهای تشخیص بیماری هاری، نقش مهمی ایفا کرد. او با بهرهگیری از بافت مغزی نمونههای انسانی، روش دقیقتری برای شناسایی این بیماری مرگبار ارائه داد. این شیوه تا دههها بهعنوان روش استاندارد باقی ماند و بار دیگر نبوغ او را اثبات کرد.
زنان در سایه، اما نه برای همیشه
ویلیامز تنها یک دانشمند نبود؛ او پیشگام فرهنگی نیز بود. در محیطهایی که زنان را جدی نمیگرفتند، آنا نهتنها خود را اثبات کرد، بلکه راه را برای دیگر زنان باز کرد. او تیمهای تحقیقاتی متشکل از زنان ایجاد کرد و نخستین زنی بود که در سال ۱۹۳۲ ریاست بخش آزمایشگاه انجمن بهداشت عمومی آمریکا را بر عهده گرفت.
نویسندهای برای آینده
آنا ویسلز ویلیامز نهفقط در آزمایشگاه، بلکه در نوشتن هم درخشید. او با همکاری ویلیام پارک، کتابهایی مانند Pathogenic Micro-organisms و Who’s Who Among the Microbes را نوشت که سالها منبع اصلی آموزش میکروبشناسی بودند.
میراثی که هنوز میدرخشد
وقتی در سال ۱۹۵۴ چشم از جهان فروبست، دنیای پزشکی زنی را از دست داد که بیهیاهو، ولی با اثری جاودانه، علم را به خدمت بشریت درآورد. امروز اگر واکسنی هست که دیفتری را مهار میکند، یا اگر آزمایشگاهی میداند چگونه بیماری را در نطفه شناسایی کند، بخشی از آن را مدیون آنا ویسلز ویلیامز هستیم.
آنا ویسلز ویلیامززنی بود که تاریخ پزشکی را بازنویسی کرد، نه با فریاد، بلکه با زمزمهای در دل آزمایشگاه.
راهکارهای کاهش ورم پا ناشی از مصرف داروی آمیلودیپین موضوع این مقاله از سایت دکتر ابوئی می باشد. آمیلودیپین یکی از داروهای متداول برای درمان فشار خون بالا و بیماریهای قلبی-عروقی است. این دارو با مسدود کردن کانالهای کلسیمی، به کاهش فشار خون کمک میکند اما یکی از عوارض جانبی رایج آن، ورم پا (ادم محیطی) است. این عارضه اغلب در اثر گشاد شدن عروق و افزایش تجمع مایعات در بافتهای پایینتنه رخ میدهد. در این مقاله، دلایل این عارضه، عوامل مؤثر بر شدت آن، و راهکارهای کاهش ورم پا مورد بررسی قرار میگیرند.
چرا آمیلودیپین باعث ورم پا میشود؟
آمیلودیپین نوعی مسدودکننده کانال کلسیمی (CCB) است که باعث گشاد شدن عروق محیطی میشود. این فرآیند فشار خون را کاهش میدهد اما در برخی افراد موجب افزایش نفوذپذیری مویرگها و تجمع مایعات در بافتها، بهویژه در پاها، میشود.
📌 دلایل اصلی این عارضه: ✅ گشاد شدن عروق محیطی → افزایش ورود مایعات به بافتهای پا ✅ کاهش بازگشت خون وریدی → مایعات در پا باقی میمانند ✅ افزایش فشار هیدرواستاتیک در مویرگها → تورم ایجاد میشود
📌 چه کسانی بیشتر در معرض ورم پا هستند؟ 🔹 افراد مسن 🔹 مبتلایان به بیماریهای قلبی یا کلیوی 🔹 کسانی که دوز بالایی از آمیلودیپین مصرف میکنند 🔹 افرادی که تحرک کمی دارند
روشهای کاهش ورم پا ناشی از آمیلودیپین
اگرچه این عارضه معمولاً خطرناک نیست، اما میتواند ناراحتکننده باشد. روشهای مختلفی برای کاهش ورم پا وجود دارد که در ادامه بررسی میشوند:
الف) تغییر دوز و جایگزینهای دارویی
✅ کاهش دوز آمیلودیپین: در برخی بیماران، کاهش دوز از ۱۰mg به ۵mg میتواند ورم را کاهش دهد بدون اینکه اثرات درمانی دارو مختل شوند. ✅ جایگزین کردن دارو: پزشک ممکن است به جای آمیلودیپین، داروهای دیگر مانند لاسارتان (Losartan) یا بتا بلاکرها را پیشنهاد دهد. این داروها تأثیر مشابهی در کنترل فشار خون دارند اما کمتر باعث ورم پا میشوند. ✅ ترکیب آمیلودیپین با دیورتیکها:داروهای مدر (دیورتیکها) مانند هیدروکلروتیازید میتوانند به دفع مایعات اضافی و کاهش تورم کمک کنند.
ب) اصلاح سبک زندگی
✅ افزایش تحرک و ورزش: حرکت دادن پاها و ورزشهای سبک مانند پیادهروی، یوگا یا شنا میتواند جریان خون وریدی را بهبود ببخشد و تجمع مایعات را کاهش دهد. ✅ بالا بردن پاها: بالا نگه داشتن پاها در هنگام استراحت باعث تخلیه مایعات اضافی از پاها میشود. توصیه میشود روزانه چندین بار پاها را بالاتر از سطح قلب قرار دهید. ✅ استفاده از جورابهای فشاری: این جورابها با افزایش فشار خارجی بر پاها، تورم را کاهش داده و از تجمع مایعات جلوگیری میکنند.
ج) تغییرات تغذیهای
✅ کاهش مصرف نمک: سدیم موجود در نمک باعث احتباس آب در بدن شده و ورم را تشدید میکند. کاهش مصرف غذاهای شور مانند فستفودها و کنسروها توصیه میشود. ✅ افزایش مصرف مواد غذایی مدر: برخی خوراکیها مانند خیار، هندوانه، جعفری و چای سبزبه دفع مایعات اضافی کمک میکنند. ✅ نوشیدن آب کافی: برخلاف تصور عمومی، نوشیدن آب کافی باعث کاهش احتباس مایعات شده و به عملکرد کلیهها کمک میکند.
د) درمانهای دارویی و طبیعی
✅ مصرف داروهای ضد التهاب: در موارد شدید، پزشک ممکن است داروهای ضد التهاب مانند ایبوپروفن تجویز کند. ✅ استفاده از ماساژ و کمپرس سرد: ماساژ پاها و استفاده از کمپرس سرد میتواند جریان خون را بهبود دهد و تورم را کاهش دهد. ✅ مکملهای مفید: برخی مطالعات نشان دادهاند که مکملهای منیزیم و پتاسیم میتوانند به تنظیم تعادل مایعات بدن کمک کنند.
چه زمانی باید به پزشک مراجعه کرد؟
در بیشتر موارد، ورم پا ناشی از آمیلودیپین مشکلی جدی نیست و با روشهای ذکر شده قابل کنترل است. اما اگر موارد زیر رخ داد، بهتر است به پزشک مراجعه کنید:
🚨 نشانههای خطرناک: ✔ تورم شدید که روزبهروز بیشتر میشود ✔ درد یا قرمزی در پاها ✔ تنگی نفس یا احساس سنگینی در قفسه سینه ✔ ورم یکطرفه (ممکن است نشانه لخته خون باشد)
پزشک ممکن است سونوگرافی، آزمایش خون یا تغییر دارو را برای کاهش این مشکل توصیه کند.
ورم پا یکی از عوارض شایع مصرف آمیلودیپین است اما معمولاً جدی نیست. با کاهش دوز دارو، تغییر سبک زندگی، رعایت رژیم غذایی و استفاده از روشهای درمانی مکمل میتوان این مشکل را مدیریت کرد. اگر ورم شدید یا همراه با علائم خطرناک باشد، باید حتماً با پزشک مشورت شود.
🔹 اگر شما هم با این مشکل مواجه هستید، تغییرات کوچک در سبک زندگی و رژیم غذایی میتواند تأثیر قابلتوجهی در بهبود شرایط داشته باشد.
📌 آیا تجربهای در این زمینه دارید؟ روشهای مؤثر شما برای کاهش تورم پا چیست؟
ویویان توماس (Vivien Thomas) یکی از چهرههای برجسته و فراموشنشدنی در تاریخ پزشکی جهان است؛ مردی که با وجود نداشتن تحصیلات دانشگاهی و قرار گرفتن در دل تبعیض نژادی، توانست یکی از مهمترین روشهای جراحی قلب را طراحی کند و جان هزاران کودک را نجات دهد. داستان او نه فقط داستان علم، بلکه داستان ایستادگی، استعداد، و انساندوستی است. در این مقاله از سایت دکتر ابوئی به بررسی این موضوع میپردازیم. در ادامه همراه ما باشید.
زندگی ابتدایی و ورود به دنیای علم
ویویان تئودور توماس در ۲۹ اوت ۱۹۱۰ در ایالت لوئیزیانای آمریکا به دنیا آمد. در دوران نوجوانی، آرزوی پزشک شدن را در سر داشت، اما وقوع رکود بزرگ اقتصادی (Great Depression) مانع ادامه تحصیل او شد. برای تأمین هزینههای زندگی، توماس به شغل نجاری روی آورد. این مهارت دستی بعدها در دقت جراحیهای او بسیار مؤثر واقع شد.
در سال ۱۹۳۰، سرنوشت مسیرش را تغییر داد؛ توماس به عنوان دستیار آزمایشگاه در دانشگاه وندربیلت استخدام شد و در آنجا با دکتر آلفرد بلالوک، یکی از چهرههای برجسته جراحی، آشنا شد. دکتر بلالوک بهسرعت استعداد توماس در کار آزمایشگاهی و درک سریع مسائل پزشکی را تشخیص داد و از او خواست تا به تیمش بپیوندد.
شاهکار علمی: درمان “سندرم کودک آبی”
یکی از مهمترین دستاوردهای ویویان توماس، همکاری با دکتر بلالوک و دکتر هلن تاسیک در طراحی روشی برای درمان “سندرم کودک آبی” بود. این بیماری یک نقص مادرزادی قلبی است که مانع از ورود خون کافی به ریهها میشود و باعث میشود پوست نوزادان به رنگ آبی درآید.
روش جراحی: شنت بلالوک-تاسیک-توماس
توماس با آزمایشهای متعدد روی سگها، موفق شد روشی نوآورانه برای اتصال سرخرگ سابکلاوین به سرخرگ ریوی طراحی کند که به خون اجازه میداد به ریهها برسد و اکسیژن کافی دریافت کند. این روش که بعدها به عنوان “شنت بلالوک-تاسیک-توماس” شناخته شد، اولین عمل موفق جراحی قلب در کودکان به حساب میآید.
در روز اولین جراحی انسانی، ویویان توماس در اتاق عمل حضور داشت و به دکتر بلالوک کمک میکرد چون او به تنهایی قادر به اجرای دقیق این تکنیک نبود. این در حالی بود که توماس هیچ مدرک پزشکی نداشت، اما مهارتش بالاتر از بسیاری از جراحان حرفهای بود.
موانع و تبعیضها
با وجود مهارتهای خارقالعادهاش، ویویان توماس سالها با حقوق اندک و عنوان شغلی پایینتر از شایستگیاش کار کرد. در مدتی از دوران کاریاش، حتی به عنوان “سرایدار” در سیستم ثبت شده بود. تبعیض نژادی گسترده مانع از به رسمیت شناخته شدن کارهای او شده بود.
اما او هیچوقت دست از تلاش برنداشت و به عنوان مربی جراحان، به بسیاری از پزشکان جوان آموزش میداد؛ پزشکانی که بعدها از جراحان مشهور آمریکا شدند.
افتخارات دیرهنگام
در نهایت، در سال ۱۹۷۶، دانشگاه جانز هاپکینز به ویویان توماس مدرک دکترای افتخاری اعطا کرد و او را به عنوان استاد جراحی منصوب نمود. این یکی از معدود دفعاتی بود که دانشگاهی معتبر، جایگاه علمی کسی را به رسمیت میشناخت که هیچ مدرک رسمی دانشگاهی نداشت.
در سالهای اخیر، نام او به طور رسمی در کنار بلالوک و تاسیک، به عنوان یکی از بنیانگذاران روش “شنت قلبی” ثبت شده است.
میراث و تأثیرات فرهنگی
زندگی ویویان توماس الهامبخش فیلمها و مستندهای زیادی شده است. فیلم تلویزیونی “Something the Lord Made” که توسط HBO ساخته شد، داستان زندگی او را با بازی Mos Def در نقش توماس به تصویر کشیده است. همچنین مستند “Partners of the Heart” توسط PBS ساخته شد و جزئیات فعالیتهای علمی و شخصی او را روایت میکند.
نتیجهگیری
ویویان توماس نمونهای زنده از این حقیقت است که استعداد، پشتکار، و نیت خیر میتواند حتی بزرگترین موانع را از سر راه بردارد. او با وجود محدودیتهای اجتماعی، اقتصادی، و آموزشی، به یکی از پایهگذاران مهم جراحی قلب مدرن تبدیل شد و نقش غیرقابل انکاری در نجات جان هزاران کودک داشت.
نام ویویان توماس، نماد توانمندی انسان در عبور از مرزهاست.
سندرم مارفان (Marfan Syndrome) یک بیماری ژنتیکی نادر اما جدی است که با درگیری گسترده بافت همبند در ارگانهای مختلف بدن از جمله سیستم اسکلتی، چشمی و قلبی-عروقی شناخته میشود. این سندرم ناشی از جهش در ژن FBN1 است که در تولید فیبریلین-۱ نقش دارد. علائم بالینی متنوع آن و تأثیر آن بر حیات بیماران، لزوم تشخیص زودهنگام و مراقبتهای چندتخصصی را ایجاب میکند. در این مقاله از سایت دکتر ابوئی به بررسی جنبههای ژنتیکی، تظاهرات بالینی، روشهای تشخیصی و درمانی سندرم مارفان میپردازیم.
مقدمه
سندرم مارفان نخستینبار توسط پزشک فرانسوی، آنتوان مارفان، در سال ۱۸۹۶ توصیف شد. این بیماری با شیوع تقریبی ۱ در ۵۰۰۰ نفر بهعنوان یک اختلال با وراثت اتوزوم غالب شناخته میشود. ژن معیوب در این بیماری، FBN1، بر روی کروموزوم ۱۵ قرار دارد و در ساخت فیبریلین-۱، پروتئینی که نقش مهمی در پایداری بافت همبند دارد، دخیل است.
ژنتیک و پاتوفیزیولوژی
ژن FBN1 کدکنندهٔ پروتئین فیبریلین-۱ است. جهش در این ژن منجر به تولید فرم ناقص این پروتئین شده و در نهایت موجب ضعف ساختاری در رشتههای میکروفیبریل میشود. این اختلال باعث انعطافپذیری بیشازحد، ضعف دیوارههای رگها (خصوصاً آئورت)، و بدشکلیهای اسکلتی میگردد.
در حدود ۷۵% موارد، بیماری از یک والد مبتلا به صورت ارثی منتقل میشود، اما در ۲۵% موارد، جهش جدید (de novo) در فرزند اتفاق میافتد.
تظاهرات بالینی
سیستم اسکلتی
قامت بلند و کشیده
بازوها و انگشتان بلند (arachnodactyly)
اسکولیوز (انحنای ستون فقرات)
قفسه سینه دفرمه (pectus excavatum یا pectus carinatum)
مفاصل بیشازحد انعطافپذیر
سیستم چشمی
دررفتگی عدسی (ectopia lentis) در حدود ۶۰-۸۰% موارد
نزدیکبینی شدید
افزایش ریسک دژنراسیون شبکیه یا جداشدگی شبکیه
سیستم قلبی-عروقی
گشادی آئورت (aortic root dilation)
آنوریسم آئورت و پارگی آن (عمدهترین علت مرگ)
نارسایی دریچه میترال
سوفل قلبی
سایر علائم
پنوموتوراکس خودبهخودی
کشیدگی پوست
فتقهای مکرر
تشخیص
تشخیص سندرم مارفان بر اساس ترکیبی از شواهد بالینی، سابقه خانوادگی و آزمایشهای ژنتیکی انجام میشود. معیارهای تشخیصی بهنام معیار گنت (Ghent Criteria) شناخته میشوند که شامل موارد زیر هستند:
گشادی ریشه آئورت
دررفتگی عدسی
جهش در ژن FBN1
سابقه خانوادگی مثبت
ابزارهای تشخیصی شامل:
اکوکاردیوگرافی
MRI یا CT آنژیوگرافی برای بررسی آئورت
معاینه چشم با اسلیت لامپ
تستهای ژنتیکی
درمان و مدیریت
درمان قطعی برای سندرم مارفان وجود ندارد، اما با مدیریت مناسب میتوان عوارض آن را کنترل کرد و طول عمر را افزایش داد.
درمان دارویی
بتابلوکرها (مثل آتنولول): کاهش فشار روی آئورت
مسدودکنندههای گیرنده آنژیوتانسین II (ARBs) مانند لوزارتان
اقدامات جراحی
تعویض ریشه آئورت در صورت گشادی شدید یا رشد سریع آن
ترمیم یا تعویض دریچه میترال در صورت نارسایی شدید
اصلاح جراحی دفورمیتیهای اسکلتی در موارد شدید
مراقبتهای دیگر
پرهیز از ورزشهای پرفشار
معاینات منظم قلب، چشم و اسکلت
مشاوره ژنتیک برای بیماران و خانوادهها
پیشآگهی
با تشخیص زودهنگام و مراقبتهای پزشکی مناسب، امید به زندگی بیماران مارفان به میزان قابل توجهی افزایش یافته است. با درمانهای محافظهکارانه و مداخلات جراحی در زمان مناسب، بسیاری از بیماران زندگی تقریباً طبیعی دارند.
سندرم مارفان، علیرغم نادر بودن، بیماریای با اهمیت بالینی بسیار است که در صورت عدم تشخیص و پیگیری مناسب میتواند تهدیدکننده حیات باشد. درمانهای نوین دارویی و پیشرفت در تکنیکهای جراحی، آیندهای امیدوارکننده برای مبتلایان فراهم کرده است. آگاهیرسانی، غربالگری ژنتیکی و پیگیریهای تخصصی نقش مهمی در بهبود کیفیت زندگی این بیماران ایفا میکنند.
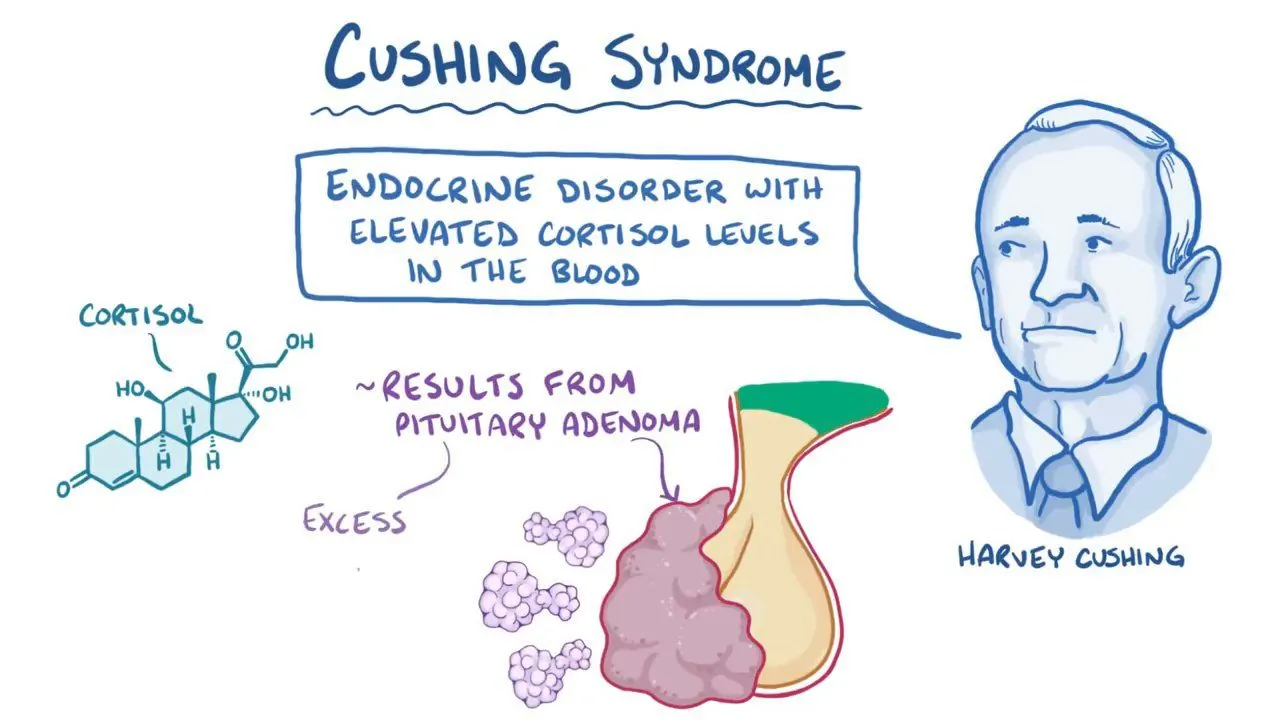
سندرم کوشینگ (Cushing’s Syndrome) یکی از بیماریهای مهم و نسبتاً نادر در حوزه غدد درونریز است که به علت افزایش مزمن سطح هورمون کورتیزول در بدن به وجود میآید. کورتیزول که به عنوان “هورمون استرس” شناخته میشود، نقش حیاتی در تنظیم متابولیسم، فشار خون، عملکرد سیستم ایمنی و پاسخ به استرس دارد. اختلال در ترشح یا مصرف این هورمون میتواند اثرات گستردهای بر سلامت فرد بگذارد. در این مقاله از سایت دکتر ابوئی به بررسی این موضوع میپردازیم.
کورتیزول چیست و چگونه عمل میکند؟
کورتیزول هورمونی است که توسط بخش قشری غده فوقکلیه (آدرنال) ترشح میشود. ترشح آن تحت کنترل محور هیپوتالاموس–هیپوفیز–آدرنال (HPA axis) قرار دارد. هیپوتالاموس هورمون CRH را ترشح میکند که هیپوفیز را به ترشح ACTH تحریک میکند. ACTH نیز باعث تحریک آدرنال برای ترشح کورتیزول میشود.
کورتیزول در شرایط استرسزا به بدن کمک میکند با افزایش قند خون، تنظیم فشار خون و مهار واکنشهای التهابی، شرایط بحرانی را مدیریت کند. اما وقتی سطح این هورمون به صورت مزمن بالا بماند، آثار مخربی بر بدن خواهد داشت.
علل بروز سندرم کوشینگ
علت اصلی این سندرم، وجود سطح بالای کورتیزول در خون است که میتواند منشأ درونی (اندوژن) یا بیرونی (اگزوجن) داشته باشد:
سندرم کوشینگ ناشی از عوامل بیرونی (شایعترین علت):
استفاده طولانیمدت و دوز بالای داروهای کورتوندار مانند پردنیزولون، دگزامتازون یا متیل پردنیزولون برای درمان بیماریهایی مثل آسم، لوپوس، یا آرتریت روماتوئید.
سندرم کوشینگ ناشی از عوامل درونی:
بیماری کوشینگ (Cushing’s Disease): شایعترین نوع سندرم کوشینگ درونی که به علت وجود تومور خوشخیم در غده هیپوفیز ایجاد میشود. این تومور باعث ترشح بیش از حد ACTH و در نتیجه افزایش کورتیزول میشود.
تومورهای آدرنال: تومورهای خوشخیم یا بدخیم غده آدرنال که بهطور مستقل کورتیزول ترشح میکنند.
کوشینگ اکتوپیک: ترشح غیرطبیعی ACTH توسط تومورهایی خارج از هیپوفیز، مانند سرطان ریه سلول کوچک، که باعث تحریک غدد فوق کلیه میشود.
علائم و نشانههای سندرم کوشینگ
علائم سندرم کوشینگ بهتدریج ظاهر میشوند و ممکن است با سایر بیماریها اشتباه گرفته شوند. مهمترین نشانهها شامل:
افزایش وزن غیرطبیعی، بهویژه در ناحیه تنه و صورت (صورت ماهمانند)
چربی در ناحیه پشت گردن (buffalo hump)
پوکی استخوان
فشار خون بالا
افزایش قند خون یا بروز دیابت نوع ۲
ضعف عضلات اندامها، بهویژه پاها
کبودی آسان و نازک شدن پوست
ترکهای پوستی بنفش یا صورتی در ناحیه شکم، ران یا سینه
اختلالات قاعدگی در زنان
کاهش میل جنسی
تغییرات خلقی مانند اضطراب، افسردگی یا تحریکپذیری
روشهای تشخیص سندرم کوشینگ
تشخیص دقیق این بیماری نیاز به بررسیهای تخصصی دارد. مهمترین روشهای تشخیصی شامل:
اندازهگیری کورتیزول در ادرار ۲۴ ساعته
آزمایش سرکوب کورتیزول با دگزامتازون
اندازهگیری کورتیزول بزاقی در نیمهشب
تصویربرداری از هیپوفیز و غدد فوق کلیوی با MRI یا CT scan
اندازهگیری ACTH برای افتراق بین انواع مختلف کوشینگ
جهت انجام آزمایشات این بیماری میتوانید به آزمایشگاه رادمان مراجعه کنید.
درمان سندرم کوشینگ
درمان بسته به علت زمینهای متفاوت است:
اگر علت مصرف داروهای کورتونی باشد:
کاهش تدریجی دوز دارو (تحت نظر پزشک) یا جایگزینی داروهای دیگر.
اگر علت تومور باشد:
جراحی: اولین انتخاب در اکثر موارد، بهویژه برای تومورهای هیپوفیز یا آدرنال.
پرتودرمانی: در صورتیکه جراحی کامل ممکن نباشد.
دارو درمانی: استفاده از داروهایی مانند کتوکونازول، میتوتان یا metyrapone برای مهار تولید کورتیزول.
برداشتن دوطرفه غدد فوق کلیه: در موارد مقاوم یا تومورهای غیرقابلجراحی.
پیشآگهی و زندگی با سندرم کوشینگ
در صورت تشخیص و درمان بهموقع، بسیاری از بیماران میتوانند به زندگی عادی بازگردند. اما در صورت عدم درمان، سندرم کوشینگ میتواند منجر به عوارض جدی مانند بیماری قلبی، دیابت و پوکی استخوان شود.
بسیاری از علائم پس از درمان بهتدریج بهبود مییابند، ولی برخی تغییرات مانند پوکی استخوان ممکن است نیاز به درمان طولانیمدت داشته باشند. همچنین پیگیری منظم توسط متخصص غدد ضروری است.
نتیجهگیری
سندرم کوشینگ یک بیماری پیچیده ولی قابل درمان است که در صورت تشخیص زودهنگام، میتوان از عوارض جدی آن جلوگیری کرد. آگاهی از علائم و پیگیری مناسب نقش کلیدی در کنترل این بیماری دارد. با همکاری بیمار و تیم درمان، امکان بازگشت به زندگی سالم کاملاً ممکن است.
لوسمی حاد میلوئید (Acute Myeloid Leukemia – AML) یکی از انواع شایع و خطرناک سرطانهای خون و مغز استخوان است که با تکثیر غیرطبیعی سلولهای نابالغ میلوئیدی در مغز استخوان مشخص میشود. این بیماری روندی تهاجمی دارد و بدون درمان سریع میتواند به سرعت پیشرفت کند. AML اغلب در بزرگسالان، بهویژه افراد بالای ۶۵ سال، دیده میشود، اما میتواند در هر سنی رخ دهد. در این مقاله از سایت دکتر ابوئی به بررسی این موضوع میپردازیم. در ادامه همراه ما باشید.
علتها و عوامل خطر لوسمی حاد میلوئید
علت دقیق AML ناشناخته است، اما برخی عوامل میتوانند خطر ابتلا را افزایش دهند:
قرار گرفتن در معرض مواد شیمیایی مانند بنزن یا داروهای شیمیدرمانی خاص
پرتودرمانی یا قرار گرفتن در معرض اشعه
اختلالات خونی پیشزمینهای مانند سندروم میلودیسپلاستیک
نقصهای ژنتیکی مانند سندروم داون
سابقه خانوادگی لوسمی (نادر است)
علائم بالینی لوسمی حاد میلوئید
علائم AML معمولاً به دلیل کاهش عملکرد مغز استخوان و افزایش سلولهای سرطانی ظاهر میشود:
خستگی مفرط و ضعف عمومی
کمخونی و رنگپریدگی
تب و عفونتهای مکرر
خونریزی یا کبودیهای غیرطبیعی (به دلیل کاهش پلاکت)
درمان AML به سن، وضعیت عمومی بیمار، و ویژگیهای ژنتیکی بیماری بستگی دارد و ممکن است شامل موارد زیر باشد:
شیمیدرمانی القایی: برای از بین بردن سلولهای سرطانی و ایجاد بهبودی اولیه
شیمیدرمانی تثبیتی یا ادامهدهنده: برای جلوگیری از عود
پیوند سلولهای بنیادی خونی (در برخی بیماران، به ویژه جوانترها یا در موارد پرخطر)
درمان هدفمند: استفاده از داروهایی که روی جهشهای خاص تأثیر میگذارند (مانند FLT3 inhibitors)
درمان حمایتی: شامل آنتیبیوتیکها، تزریق خون و مراقبتهای تغذیهای
پیشآگهی و عود بیماری
پیشآگهی AML بستگی زیادی به سن بیمار، نوع ژنتیکی سرطان، پاسخ به درمان اولیه، و وجود سایر بیماریهای زمینهای دارد. نرخ بقای پنجساله برای بزرگسالان حدود ۳۰٪ است، اما در بیماران جوانتر و دارای شرایط مطلوبتر، این رقم میتواند بسیار بالاتر باشد.
جمعبندی
لوسمی حاد میلوئید یک بیماری جدی و نیازمند درمان فوری است. پیشرفتهای پزشکی در زمینه درمان هدفمند، پیوند سلولهای بنیادی، و تکنولوژیهای تشخیصی، امیدهای تازهای برای بیماران به همراه داشتهاند. غربالگری منظم در بیماران پرخطر و تشخیص زودهنگام میتواند نقش کلیدی در افزایش شانس موفقیت درمان ایفا کند.
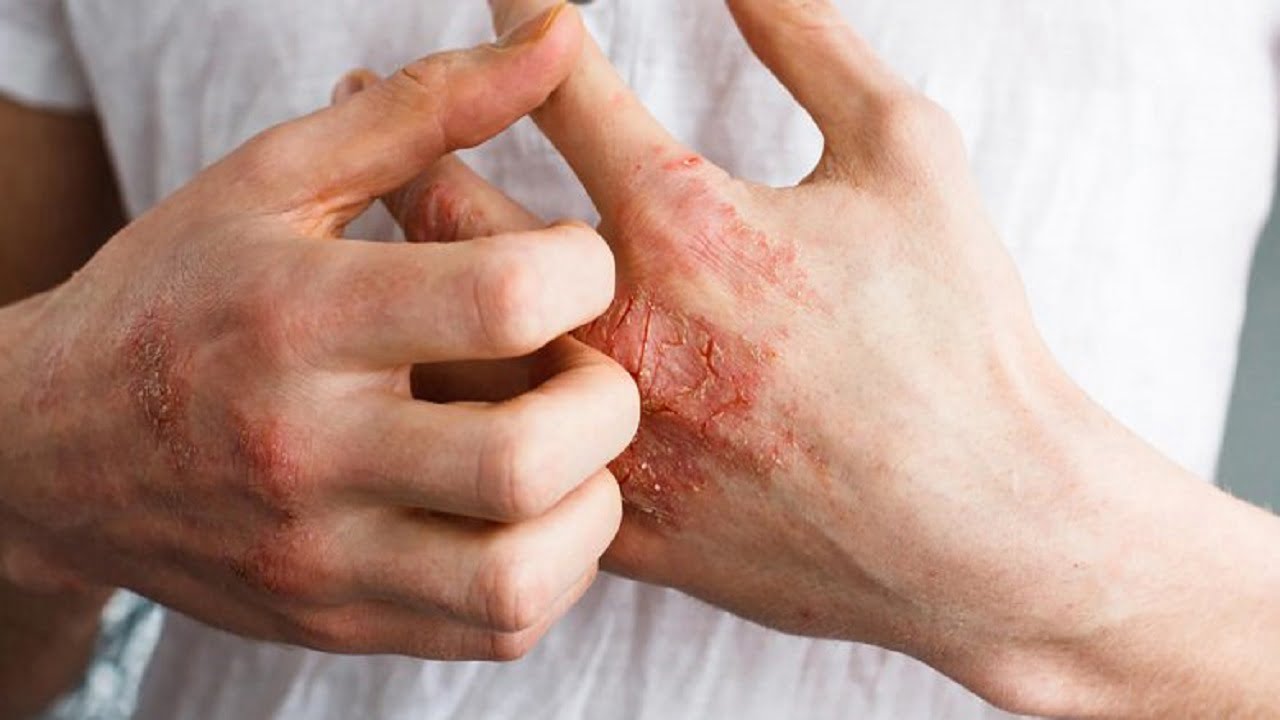
خارش (Pruritus) یک احساس ناخوشایند در پوست است که منجر به تمایل به خاراندن میشود. این علامت ممکن است بهتنهایی یا به همراه ضایعات پوستی ظاهر شود و میتواند علامت بیماریهای پوستی، سیستمیک، عصبی یا روانی باشد. شناخت منشاء خارش برای درمان موفق آن حیاتی است. در این مقاله از سایت دکتر ابوئی به بررسی این موضوع میپردازیم. در ادامه همراه ما باشید.
خارش با منشأ پوستی (Dermatologic Itch)
این نوع خارش شایعترین نوع است و معمولاً با ضایعات پوستی قابل مشاهده همراه است.
الف. اگزما (Atopic Dermatitis)
شرح حال: معمولاً از کودکی آغاز میشود؛ همراه با سابقه آتوپی در خانواده.
علائم: خشکی پوست، قرمزی، پوستهریزی، ترک، و خارش شدید. نواحی شایع: چینها (پشت زانو، داخل آرنج).
جهت مشاهده بهتر جدول در تلفن همراه، آن را در حالت افقی قرار دهید
منشأ
درمان
پوستی (اگزما، گال…)
کورتون موضعی، آنتیهیستامین، ضدقارچ
کبدی
کلستیرامین، اورسودئوکسیکولیک اسید
کلیوی
دیالیز بهینه، نوردرمانی
نورولوژیک
گاباپنتین، پرهگابالین، TCA
روانزاد
SSRIs، رواندرمانی
عمومی
مرطوبکننده، حمام جو دوسر، آنتیهیستامین نسل دوم
روش افتراق تشخیصی خارش: فرآیند سیستماتیک پزشک برای شناسایی علت اصلی
فرآیند تشخیص افتراقی خارش نیازمند یک بررسی گامبهگام کلینیکی است. پزشک از تاریخچهگیری دقیق، معاینه فیزیکی جامع، و سپس انجام تستهای آزمایشگاهی و تصویربرداری (در صورت نیاز) برای تمایز بین علل پوستی، سیستمیک، نوروژنیک و سایکوژنیک استفاده میکند.
مرحله اول: آنالیز تاریخچه بالینی (History Taking)
پزشک ابتدا با سؤالاتی دقیق به جمعآوری اطلاعات کلیدی میپردازد:
✅سوالات مهم:
مدت زمان خارش: حاد (<6 هفته) یا مزمن (≥۶ هفته)
الگوی زمانی: شبانه (Scabies، Cholestasis)، پس از حمام (Aquagenic pruritus)، فصلی
مرد ۲۳ ساله، خارش در ناحیه تناسلی و بین انگشتان، بدتر در شب
همسرش هم علائم دارد
تشخیص: گال
درمان: پرمترین، درمان کل خانواده
نتیجهگیری تکمیلی
خارش یک نشانه بسیار متنوع است که ممکن است از یک اختلال ساده پوستی تا یک بیماری تهدیدکننده حیات مانند لنفوم ناشی شده باشد. بررسی دقیق بالینی و انجام آزمایشهای هدفمند برای تشخیص دقیق، و انتخاب درمان بر اساس علت زمینهای، در مدیریت صحیح خارش بسیار اهمیت دارد. در مدیریت خارش، پزشک نقش کارآگاهی دارد که باید با ترکیب دقیق شرح حال، معاینه، و بررسیهای پاراکلینیکی، علت اصلی را شناسایی کند. در صورت نبود ضایعه پوستی، ذهن پزشک باید به سمت علل سیستمیک، نورولوژیک و روانی سوق داده شود. تشخیص افتراقی ساختارمند، کلید درمان مؤثر و جلوگیری از مزمن شدن خارش است.