
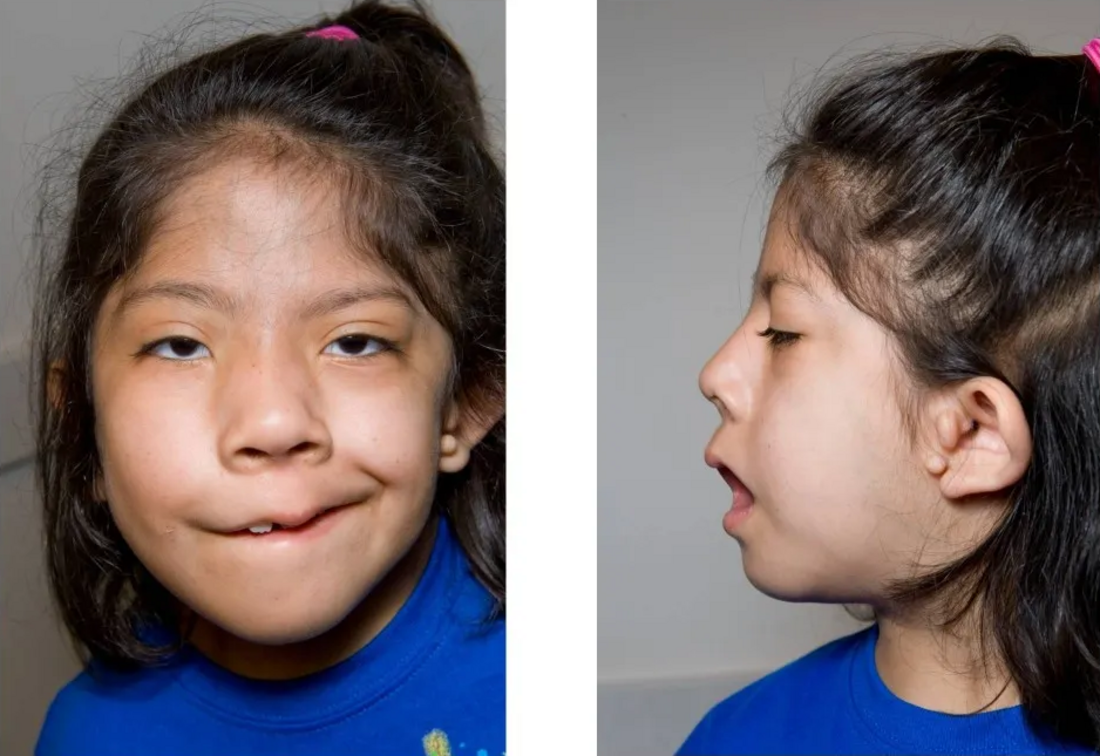
سندرم گلدنهار که با نام Oculo-Auriculo-Vertebral Spectrum نیز شناخته میشود، یک اختلال مادرزادی نادر است که بهطور عمده بر رشد ساختارهای صورت، گوش، چشم و ستون فقرات تأثیر میگذارد. این سندرم بخشی از طیف وسیعی از اختلالات کرانیوفاشیال است و در نتیجه نقص در تکامل قوسهای برانشیال (Branchial Arches) در دوران جنینی پدید میآید. در این مقاله از سایت دکتر ابوئی به بررسی این موضوع میپردازیم. در ادامه این مقاله همراه ما باشید.
اپیدمیولوژی
شیوع سندرم گلدنهار تقریباً ۱ در هر ۳,۰۰۰ تا ۵,۰۰۰ تولد زنده است. این بیماری ممکن است در هر دو جنس دیده شود، اما در برخی مطالعات، شیوع آن در پسران کمی بیشتر گزارش شده است. بیشتر موارد بهصورت اسپورادیک (غیروارثی) رخ میدهند، اگرچه موارد خانوادگی نیز گزارش شدهاند.
علل و پاتوفیزیولوژی
علت دقیق سندرم گلدنهار هنوز ناشناخته است، اما تحقیقات نشان دادهاند که این بیماری در اثر اختلال در رشد قوسهای اول و دوم برانشیال در مراحل اولیه رشد جنینی (هفته چهارم تا هشتم بارداری) ایجاد میشود. عوامل ژنتیکی، اپیژنتیکی، و محیطی از جمله عفونتهای ویروسی، مصرف داروهای تراتوژنیک، دیابت بارداری، و کمبود جریان خون جفتی میتوانند در ایجاد آن نقش داشته باشند.
تظاهرات بالینی
علائم سندرم گلدنهار بهصورت طیفی از خفیف تا شدید بروز میکنند و ممکن است یکطرفه یا دوطرفه باشند. در ادامه، شایعترین تظاهرات این بیماری آمده است:
ناهنجاریهای چهرهای
- عدم تقارن صورت (Hemifacial microsomia)
- کوچکی یا عدم رشد فک پایین (ماندیبل)
- کام شکری یا شکاف لب
- کجی بینی یا فک بالا
مشکلات گوش
- میکروتیا (کوچکی گوش)
- آنوتیا (عدم وجود گوش)
- سوراخ یا زائدههای پیشگوشی
- ناشنوایی انتقالی یا حسیعصبی
مشکلات چشمی
- درمالیپوم (Lipodermoid) یا چربی زیر ملتحمه
- کاهش عملکرد عضلات چشم
- ناهنجاریهای پلک یا موقعیت چشم
ناهنجاریهای مهرهای و ستون فقرات
- همیورتبرا
- اسکولیوز (انحنای جانبی ستون فقرات)
- فیوژن مهرهای
اختلالات سیستمیک
- نقایص قلبی: مانند نقص دیواره بین دهلیزی یا بطنی
- نقایص کلیوی: کلیه نعل اسبی، کلیه نابجا
- اختلالات عصبی: شامل هیدروسفالی، صرع، یا تأخیر ذهنی در برخی موارد
تشخیص
تشخیص سندرم گلدنهار عمدتاً بالینی و مبتنی بر مشاهده ویژگیهای ظاهری در بدو تولد است. ابزارهای کمکی عبارتاند از:
- تصویربرداری: CT Scan و MRI برای بررسی استخوانهای صورت و جمجمه
- اشعه X ستون فقرات برای بررسی ناهنجاریهای مهرهای
- آزمایشهای شنوایی و بینایی
- اکوکاردیوگرافی برای ارزیابی قلب
- سونوگرافی کلیهها
- آزمایشهای ژنتیک در صورت نیاز
آشنایی با آزمایشگاه رادمان فردیس
افتراق تشخیصی
- سندرم ترچر کالینز (Treacher Collins Syndrome)
- سندرم ناجر (Nager Syndrome)
- میکروزومیا همیفاشیال غیرسندرومی
- سندرم CHARGE
درمان
درمان سندرم گلدنهار نیازمند یک رویکرد چندتخصصی و بلندمدت است که معمولاً شامل مراحل زیر میشود:
درمانهای جراحی
- بازسازی گوش (اتولوژی یا پروتز)
- جراحی فک و صورت (ارتوگناتیک)
- اصلاح شکاف لب و کام
- اصلاح اسکولیوز یا مشکلات مهرهای
درمانهای توانبخشی
- گفتاردرمانی
- کاردرمانی
- شنواییشناسی و تجویز سمعک در صورت نیاز
حمایت روانی و اجتماعی
- آموزش والدین
- مشاوره ژنتیک
- حمایت تحصیلی و شناختی در صورت وجود مشکلات یادگیری
پیشآگهی
پیشآگهی سندرم گلدنهار بسته به شدت تظاهرات و درگیری ارگانها متفاوت است. در اغلب موارد، با مداخلات پزشکی و جراحی مناسب، کیفیت زندگی بیماران بهطور قابل توجهی بهبود مییابد. موارد شدید با اختلالات متعدد ممکن است نیازمند پیگیری مادامالعمر باشند.
نتیجهگیری
سندرم گلدنهار یک اختلال پیچیده و متغیر است که نیاز به تشخیص زودهنگام، مداخلات چندرشتهای و پیگیری منظم دارد. آگاهی از این بیماری در میان پزشکان، والدین و متخصصان رشد کودک میتواند نقش بسزایی در بهبود کیفیت زندگی مبتلایان ایفا کند.
منابع:
- Rollnick, B.R., et al. (1987). Oculoauriculovertebral Spectrum: An Updated Review. Journal of Craniofacial Genetics and Developmental Biology.
- Tasse, C., et al. (2005). Phenotypic spectrum of the Oculo-auriculo-vertebral spectrum: A review of 74 patients. American Journal of Medical Genetics.
- Online Mendelian Inheritance in Man (OMIM): Goldenhar Syndrome.