
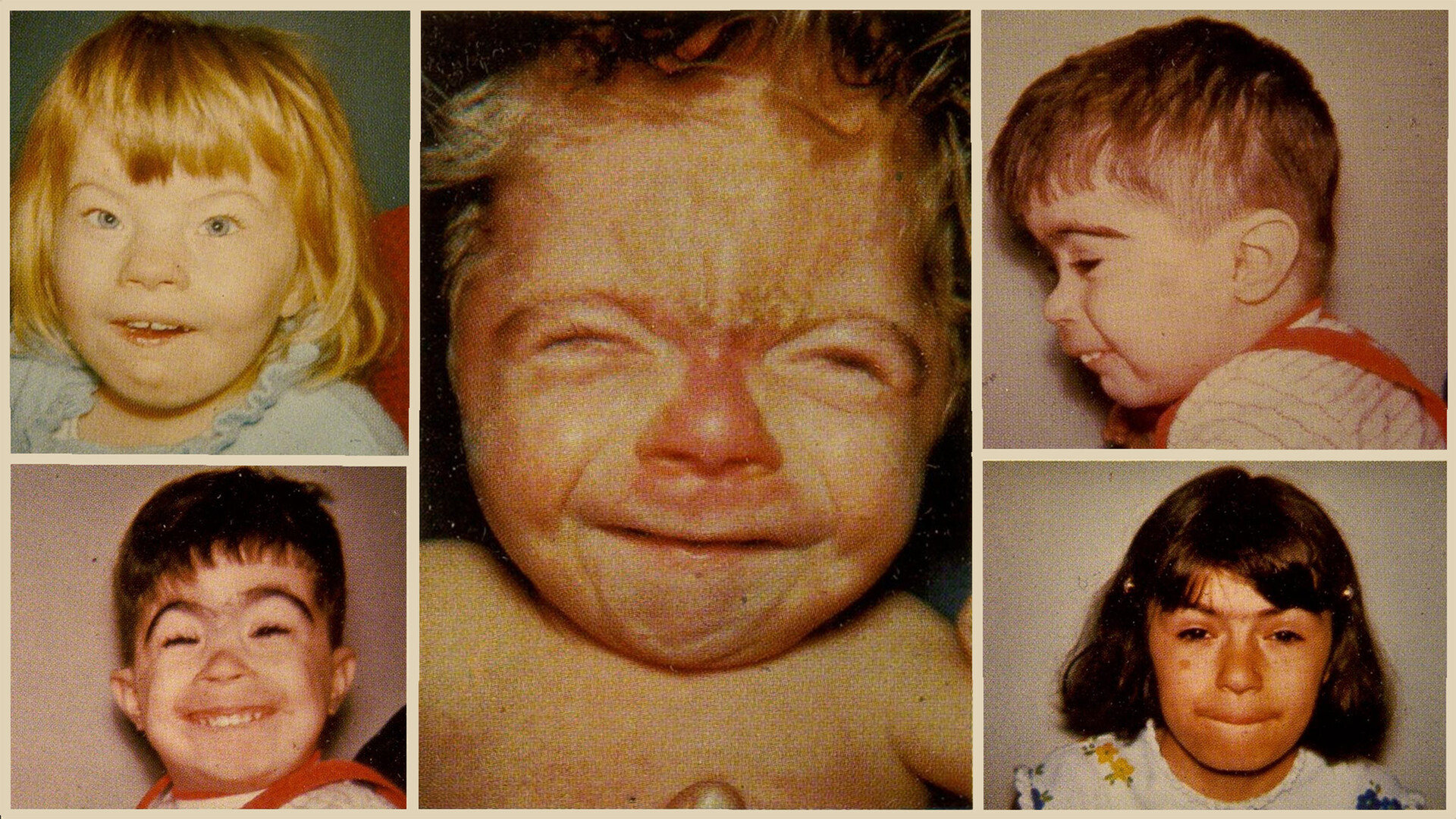
سندرم کورنیلا د لانگ (Cornelia de Lange Syndrome – CdLS) یک اختلال ژنتیکی نادر است که رشد فیزیکی، ذهنی و رفتاری را تحت تأثیر قرار میدهد. این سندرم نخستین بار توسط دکتر کورنیلا د لانگ، متخصص کودکان هلندی، در سال ۱۹۳۳ توصیف شد. میزان بروز آن تخمین زده میشود که بین ۱ در هر ۱۰٬۰۰۰ تا ۳۰٬۰۰۰ تولد زنده باشد. در این مقاله از سایت دکتر ابوئی به سندرم سندرم کورنیلا د لانگ میپردازیم، در ادامه همراه ما باشید.
ویژگیهای بالینی سندرم کورنیلا د لانگ (Clinical Features)
ویژگیهای صورت:
- سینوفریز (Synophrys): بههم پیوستن ابروها در خط میانی.
- مژههای بلند و ضخیم.
- بینی کوتاه با سوراخهای بینی به سمت پایین.
- لب بالایی نازک، لب پایینی برجسته.
- فک پایین کوچک (Micrognathia).
- گوشهای پایینتر از محل معمول و فرم غیرطبیعی.
تأخیر رشد:
- رشد داخل رحمی (Intrauterine Growth Retardation).
- قد و وزن کمتر از صدکهای طبیعی بعد از تولد.
ناهنجاریهای اندام:
- کوتاهی اندام فوقانی (مخصوصاً بازو یا ساعد).
- انگشتان اضافی (Polydactyly) یا فقدان انگشت (Oligodactyly).
- مفاصل سفت یا محدودیت حرکتی.
اختلالات ذهنی و رفتاری:
- تأخیر در گفتار.
- درجات مختلف عقبماندگی ذهنی (از خفیف تا شدید).
- رفتارهای اوتیسممانند (Autistic-like behaviors).
مشکلات سیستمیک:
- ناهنجاریهای قلبی (مانند نقص دیواره بین بطنی – VSD).
- ناهنجاریهای کلیوی.
- رفلاکس معدهبهمری (GERD).
- مشکلات شنوایی (کمشنوایی انتقالی یا عصبی-حسی).
- مشکلات بینایی (مانند نزدیکبینی یا تنبلی چشم).
علتشناسی (Etiology)
CdLS ناشی از جهش در ژنهایی است که در چسبندگی کروماتین و تنظیم رونویسی ژنتیکی دخیل هستند:
جهت مشاهده بهتر جدول در گوشی همراه، آن را به حالت افقی نگه دارید
| ژن درگیر | ویژگی | نوع وراثت |
|---|---|---|
| NIPBL (بیشتر موارد) | پروتئینی که در کمپلکس Cohesin نقش دارد | اتوزومال غالب |
| SMC1A | کروموزوم X | وابسته به X |
| SMC3 | کمپلکس Cohesin | اتوزومال غالب |
| RAD21 | ارتباط با Cohesin | اتوزومال غالب |
| HDAC8 | کنترل ساختار کروماتین | وابسته به X |
در اکثر موارد، این جهشها de novo هستند (یعنی به صورت جدید در فرد ایجاد شدهاند و از والدین به ارث نرسیدهاند).
تشخیص سندرم کورنیلا د لانگ (Diagnosis)
تشخیص مبتنی بر سه محور است:
معاینه بالینی
بر اساس ظاهر فیزیکی (صورت مشخصه، ناهنجاریهای اندام، تأخیر رشد).
آزمایشهای ژنتیکی
- توالییابی نسل جدید (NGS) یا آزمایش پنل ژنتیکی برای شناسایی جهشهای NIPBL، SMC1A، SMC3، RAD21 و HDAC8.
- در صورت شک بالینی قوی اما تست ژنتیک منفی، میتوان از آزمایشهای MLPA یا Array CGH برای کشف حذفهای کوچک (microdeletions) استفاده کرد.
سایر ارزیابیهای تکمیلی
- اکوکاردیوگرافی برای بررسی مشکلات قلبی.
- بررسیهای شنوایی و بینایی.
- تصویربرداری MRI مغز در صورت وجود اختلالات عصبی.
پیشگیری از سندرم کورنیلا د لانگ (Prevention)
مشاوره ژنتیک (Genetic Counseling)
- اگرچه بیشتر موارد CdLS تصادفی هستند، در مواردی که والدین حامل جهش باشند، ریسک تکرار وجود دارد.
- آزمایش ژنتیکی والدین در صورتی که فرزندی با CdLS داشته باشند توصیه میشود.
غربالگری پیش از تولد
- در خانوادههای با سابقه CdLS و جهش شناخته شده، میتوان با آمنیوسنتز یا نمونهبرداری از پرزهای کوریونی (CVS)، DNA جنین را آزمایش کرد.
- در سونوگرافیهای دقیق (Anomaly Scan)، نشانههایی مانند تأخیر رشد جنینی، ناهنجاری اندام فوقانی و ویژگیهای غیرطبیعی صورت ممکن است قابل تشخیص باشند، ولی قطعی نیستند.
آشنایی با آزمایشگاه رادمان فردیس
درمان و مراقبت (Management)
درمان قطعی
درمان قطعی برای CdLS وجود ندارد. درمان بر مدیریت علائم متمرکز است:
مراقبتهای پزشکی:
- درمان مشکلات گوارشی (مانند GERD با دارو یا جراحی).
- مداخلات شنوایی (سمعک یا جراحی).
- اصلاح ناهنجاریهای ارتوپدی در صورت نیاز.
توانبخشی:
- گفتاردرمانی، کاردرمانی و فیزیوتراپی برای ارتقای مهارتهای حرکتی و ارتباطی.
- درمانهای روانشناختی و رفتاری به خصوص برای مشکلات اوتیسمی و پرخاشگری.
مداخلات آموزشی:
- آموزشهای ویژه متناسب با سطح یادگیری کودک.
- پشتیبانی در محیطهای مدرسه با استفاده از مربیان آموزشدیده.
پیشآگهی (Prognosis)
بسته به شدت علائم، پیشآگهی متفاوت است. در موارد خفیفتر، افراد میتوانند زندگی نسبتاً مستقلی داشته باشند، در حالی که در موارد شدید، نیاز به مراقبتهای مادامالعمر دارند. امید به زندگی معمولاً کاهش نمییابد مگر در صورت وجود مشکلات شدید قلبی یا ریوی درماننشده.
جمعبندی
سندرم کورنیلا د لانگ یک اختلال چندسیستمی با طیف بالینی گسترده است که نیازمند تشخیص زودهنگام، مراقبت چندتخصصی، و پشتیبانی خانوادگی قوی است. با پیشرفتهای ژنتیکی در شناسایی و تشخیص، امکان غربالگری قبل از تولد نیز فراهم شده، هرچند پیشگیری از جهش de novo هنوز ممکن نیست.
سلام ببخشید دختر دایی من مشکوک به این سندرم کورنیلا دی لانگ هست و پدرش هم ناقل جا به جایی کروموزومی بود و ازدواج فامیلی داشت، میخواستم بدونم من که باردارم و سونوگرافی NTوNBرفتم نرمال بود غربالگری اول و دوم رفتم نرمال بود، سونو آنومالی دادم نرمال بود و اکوی قلب هم جنین هم رفتم نرمال بود آمنیوسنتز هم دادم جنین هم جابه جایی داره مثل خودم تو کروموزوم ۴ و ۱۸ عین کاریوتایپ خودم این جواب کاریوتایپ خودم:
.۴۶XX,t(4;18)(p16.1;q23)
این هم جواب کاریوتایپ جنین
که به این صورت نوشته :
.۴۶XX,t(4;18)(p16.1;q23)mat حالا با توجه به این حالت جنین سالم است؟ ممنون میشم راهنمایی بفرمایید.،میخواستم بدونم از کجا میتونم مطمئن بشم جنینم از لحاظ ابتلا به این سندرم کورنیلا دی لانگ سالم هستش؟ از کجا بفهمم جنینم کاملا سالم؟ سندرم کورنیلا دی لانگ مربوط به جهش در کدام کروموزوم؟
جابجاییای که تو کاریوتایپ شما و جنین دیده شده متعادل هست و مثل خودتون باعث بیماری نمیشه. سندرم کورنیلا دی لانگ به این جابجایی ربطی نداره و معمولاً به جهش ژن NIPBL روی کروموزوم ۵ (یا ژنهای مشابه) مربوط میشه. از نظر سونو و غربالگری هم که نرمال بوده. برای اطمینان کامل فقط باید تست ژنتیکی اختصاصی برای CdLS انجام بشه
اون تست اختصاصی Cdls چگونه تستی است؟
تست اختصاصی سندرم کورنیلا دی لانگ (CdLS)، بررسی ژنهای مرتبط با این سندرم مثل NIPBL و چند ژن دیگر است و معمولاً با روش توالییابی ژن یا پنل ژنتیک انجام میشود. این تست مشخص میکند آیا جهش ایجادکننده CdLS در جنین وجود دارد یا نه.
ببخشید این تست هم از طریق آمنیوسنتز انجام میشه؟ یعنی نمونه مایع امنیوتیک ازمایش میشه؟ الان ۲۱ هفته و چند روز هستم یعنی نیاز این تست برم.
ببخشید این تست هم از طریق آمنیوسنتز صورت میگیره ؟ از طریق مایع آمنیوتیک؟
سلام ببخشید آیا فقط ازدواج فامیلی باعث جهش های تک ژنی در جنین میشه؟ یا ازدواج با غریبه هم باعث جهش های تک ژنی در جنین میشه؟ آیا سندرم کورلینا دی لانگ جزو جهشهای تک ژنی است و فقط در ازدواج فامیلی این سندرم ایجاد میشه؟دختر دایی من مشکوک به این سندرم چون دایی من ازدواج فامیلی داره و جابه جایی کروموزومی داره دخترش مشکوک به سندرم کورلینا دی لانگ، من خودم هم جا به جایی کروموزومی دارم ولی با غریبه ازدواج کردم و سونو NTوNBمن نرمال بود غربالگری هام نرمال بود سونو آنومالی نرمال بود اکوی قلب جنین هم نرمال بود آمنیوسنتز دادم جوابش عین کاریوتایپ خودم جنین مثل خودم در کروموزوم ۴ و ۱۸ جابه جایی داره، حالا میخوام بدونم مطمئن باشم که جنینم کاملا سالم؟ مطمئن باشم که جنینم از لحاظ ابتلا به این سندرم کورلینا دی لانگ و جهشای ژنی سالم؟
سلام
با تشکر از کامنت شما در ادامه پاسخی مرحله ای برایتان ادامه نموده ایم.
جهشهای تک ژنی و ازدواج فامیلی:
ازدواج فامیلی ریسک برخی بیماریهای اتوزوم مغلوب تک ژنی را افزایش میدهد، چون احتمال اینکه هر دو والد ژن ناقص مشابه داشته باشند بیشتر است.
ازدواج با غیر فامیلی هم میتواند منجر به جهشهای تک ژنی شود، ولی احتمال وقوع کمتر است، چون ژنهای ناقص مشابه در والدین نادرتر هستند.
بنابراین جهش تک ژنی فقط مخصوص ازدواج فامیلی نیست، اما ازدواج فامیلی ریسک را بالاتر میبرد.
سندرم کورلینا دی لانگ:
این سندرم جزو بیماریهای ژنتیکی است که معمولاً به علت جهش تک ژنی یا تغییرات کروموزومی خاص ایجاد میشود.
در ازدواج فامیلی شانس بروز آن بیشتر است، ولی ممکن است در غیر فامیلی هم رخ دهد، اگر جهش یا تغییر کروموزومی منتقل شود.
وضعیت جنین شما:
شما گفتید کاریوتایپ جنین مشابه خودتان است (با همان جابهجایی کروموزومی)، و همه آزمایشها (سونو NT/NB، اکوی قلب، غربالگری) نرمال بودند.
این به این معنی است که جنین شما از نظر ساختار کلی و بسیاری از بیماریهای شایع کروموزومی سالم است.
با این حال، برخی سندرمهای تک ژنی و بیماریهای نادر ممکن است در کاریوتایپ مشخص نشوند و فقط با تست ژنتیکی دقیقتر (مثل توالییابی ژن یا پنل ژنتیک) قابل شناسایی باشند.
جمعبندی نهایی برای آرامش:
جنین شما با توجه به نتایج سونو و کاریوتایپ، احتمال ابتلا به بیماریهای کروموزومی جدی پایین است.
احتمال وقوع سندرمهای تک ژنی بسیار نادر و معمولاً تصادفی است، به خصوص وقتی والدین غیر فامیلی هستند و ژنهای ناقص مشابه ندارند.
اگر بخواهید اطمینان بیشتری داشته باشید، میتوان مشاوره ژنتیک پیشرفته و تست توالی ژن یا پنل سندرمها انجام داد.
ببخشید اینکه عرض کردم دختر داییم مشکوک به سندرم کورنلیا دی لانگ هستش و داییم با همسرش فامیل هست ،ازدواج فامیلی داییم تا اونجایی که میدونم با نوه ی دختر خاله اش ازدواج کرده البته دقیق نمیدونم ولی اینو میدونم که فامیل خیلی دور میخواستم بدونم علاوه بر اینکه داییم جا به جایی کروموزومی داره این ازدواجش با فامیل دور هم میتونه باعث جهش ژنی و باعث سندرم کورنلیا دی لانگ بشه ؟
سلام ببخشید آیا فقط ازدواج فامیلی باعث جهش های تک ژنی در جنین میشه؟ یا ازدواج با غریبه هم باعث جهش های تک ژنی در جنین میشه؟ آیا سندرم کورنلیا دی لانگ جزو جهشهای تک ژنی است و فقط در ازدواج فامیلی این سندرم ایجاد میشه؟دختر دایی من مشکوک به این سندرم چون دایی من ازدواج فامیلی داره و جابه جایی کروموزومی داره دخترش مشکوک به سندرم کورنلیا دی لانگ، من خودم هم جا به جایی کروموزومی دارم ولی با غریبه ازدواج کردم و سونو NTوNBمن نرمال بود غربالگری هام نرمال بود سونو آنومالی نرمال بود اکوی قلب جنین هم نرمال بود آمنیوسنتز دادم جوابش عین کاریوتایپ خودم جنین مثل خودم در کروموزوم ۴ و ۱۸ جابه جایی داره، حالا میخوام بدونم مطمئن باشم که جنینم کاملا سالم؟ مطمئن بشم که جنینم از لحاظ ابتلا به سندرم کورنلیا دی لانگ سالم؟ مطمئن باشم جنیم از لحاظ جهشهای ژنی سالم است؟
در ادامه سوالی که عرض کردم سونوگرافی NTو NBنرمال بود،آزمایشهای غربالگری اول و دوم نرمال بود، سونوی آنومالی نرمال بود و اکوی قلب جنین هم نرمال بود آمنیوسنتز هم دادم جنین مثل خودم جابه جایی کروموزومی دارد، این جواب کاریوتایپ خودم:
.۴۶XX,t(4;18)(p16.1;q23)
این هم جواب آمنیوسنتز و جواب کاریوتایپ جنین
که به این صورت نوشته :
.۴۶XX,t(4;18)(p16.1;q23)mat حالا با توجه به این حالت جنین سالم است؟ مطمئن باشم جنینم از لحاظ سندرم کورنلیا دی لانگ سالم است؟ با توجه به اینکه دختر داییم مشکوک به سندرم کورنلیا دی لانگ نگرانم میخواستم بدونم جنینم از لحاظ این سندرم سالم؟ در حالت کلی جنینم از لحاظ جهشهای تک ژنی و جهشهای ژنی سالم؟ من ازدواج فامیلی نکردم،داییم با فامیل ازدواج کرده و خودش هم جابه جایی کروموزومی داره دخترش مشکوک به سندرم کورنلیا دی لانگ هست،ممنون میشم راهنمایی بفرمایید.
سلام ببخشید در ادامه سوالم باید عرض کنم که دایی من جابه جایی کروموزومی داره و ازدواج فامیلی داره ، دخترش مشکوک به سندرم کورنلیا دی لانگ، اون یکی داییم هم بچه اش مشکل جسمی و دهنی و حرکتی داره به نظرم ژنتیکی البته دقیق نمیدونم چه مشکلی داره اونم ازدواج فامیلی داشته و احتمالا اون داییم هم ناقل جا به جایی کروموزومی هستش، من خودم هم جا به جایی کروموزومی دارم درکروموزوم ۴ و ۱۸، قبلا هم یک سقط در ۸ هفته داشتم رشد جنین متوقف شده بود در ۱۱ هفته فهمیدم جنین نبض نداره و رشدش تو ۸ هفته مونده بود ،یک سقط قانونی در ۱۸ هفته داشتم که جنین در بخش p16.1کروموزوم ۴ دچار حذف شده بود ،من با غریبه ازدواج کردم، این دفعه هم باردار هستم سونو NTوNBمن نرمال بود،آزمایشهای غربالگری هام نرمال بود، سونو آنومالی اسکن سه بعدی رفتم نرمال بود، اکوی قلب جنین هم نرمال بود، آمنیوسنتز دادم جوابش عین کاریوتایپ خودم جنین مثل خودم در کروموزوم ۴ و ۱۸ جابه جایی داره این جواب کاریوتایپ خودم:
.۴۶XX,t(4;18)(p16.1;q23)
این هم جواب کاریوتایپ آمنیوسنتز جنین
که به این صورت نوشته :
.۴۶XX,t(4;18)(p16.1;q23)mat حالا با توجه به این جواب آمنیوسنتز جنینم کاملا سالم است؟ مطمئن بشم که جنینم از لحاظ ابتلا به سندرم کورنلیا دی لانگ سالم؟ مطمئن باشم جنینم از لحاظ جهشهای ژنی و جهشای تک ژنی سالم است؟
جابجایی کروموزومی که در شما و جنین دیده شده متعادل است و باعث بیماری نمیشود. سندرم کورنلیا دی لانگ معمولاً به جهش در ژن NIPBL روی کروموزوم ۵ (و گاهی چند ژن دیگر) مربوط است و ربطی به این جابجایی ندارد.
با توجه به اینکه سونوگرافیها، غربالگریها و اکوی قلب نرمال بودهاند، ریسک بیماریهای کروموزومی جدی پایین است. برای اطمینان کامل از ابتلا به CdLS یا سایر جهشهای تک ژنی، فقط تست ژنتیکی اختصاصی روی ژنهای مرتبط با CdLS میتواند آن را مشخص کند.
به طور کلی، با این نتایج، احتمال اینکه جنین سالم باشد خیلی بالا است، اما هیچ تستی جز آزمایش ژنتیکی اختصاصی نمیتواند صددرصد تضمین کند.
ببخشید اینکه عرض کردم دختر داییم مشکوک به سندرم کورنلیا دی لانگ هستش و داییم با همسرش فامیل هست ،ازدواج فامیلی داییم تا اونجایی که میدونم با نوه ی دختر خاله اش ازدواج کرده البته دقیق نمیدونم ولی اینو میدونم که فامیل خیلی دور میخواستم بدونم علاوه بر اینکه داییم جا به جایی کروموزومی داره این ازدواجش با فامیل دور هم میتونه باعث جهش ژنی و باعث سندرم کورنلیا دی لانگ بشه ؟